IGFBP3 Research
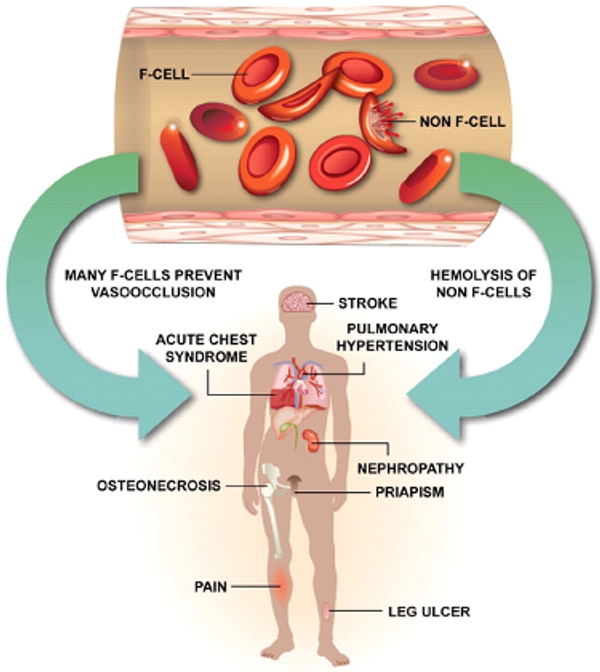
Our lab is investigating the crucial role of Insulin-like Growth Factor Binding Protein 3 (IGFBP3) in fetal hemoglobin (HbF) induction as a therapeutic approach for sickle cell disease.
This research stems from whole genome sequencing studies that revealed individuals with higher IGFBP3 levels naturally produce more fetal hemoglobin. This research provides a promising new therapeutic avenue for sickle cell disease treatment by leveraging the body's natural mechanisms for producing healthier hemoglobin.
- Whole genome sequencing identified IGFBP3 as a regulator of HbF production
- Plasma levels of IGFBP3 correlate with HbF production in patient samples
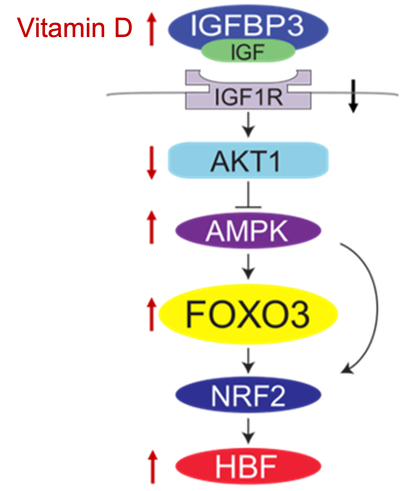
- Adding IGFBP3 to erythroid precursors increases HbF production, as verified in vitro
- IGFBP3 can be targeted therapeutically for SCD treatment
- Vitamin D increases IGFBP3 levels, offering a potential intervention strategy
- IGFBP3 deficiency is common in SCD patients
- IGFBP3-based therapies can be administered in combination with hydroxyurea